Reaction-Diffusion Systems¶
In reaction-diffusion systems, reaction processes are defined
per location for a finite set of N locations that are connected
via links in a network. Through these links, reactions between
locations can take place. This means that if we assume a reactive
system of C compartments per location, the system can be described
using an instance of epipack.numeric_matrix_epi_models.MatrixEpiModel
with \(N\times C\) compartments: Each compartment is identified
by a location and its epidemiological description.
Note
Usually, reaction diffusion equations are somewhat computationally
heavy, which is why for research purposes you should consider
other solutions or writing custom code. Nevertheless, epipack's
philosophy of fast prototyping comes in handy, so if you just aim
at finding the right model and visualize it in a small system,
the following tutorial might help.
Networks¶
One such example might be an SIR process taking place in different locations on earth which are connected via the air traffic network. In this case, the reaction equations read
Here, \(X_u\) represents a compartment \(X\in\{S,I,R\}\) at location \(u\) with population size \(N_u\), and \(w_{uv}\) represents a transition rate of an individual from location u to location v.
In this picture, the total number of individuals traveling in the network remains constant and it is assumed that the population size per location is constant for all nodes, as well, if \(w_{uv}=w_{vu}\). The last equation describes an edge-centric random walk, which is associated with a uniform equilibrium concentration.
Let's assume we have an undirected network \(A_{uv}\) and the transition rate per link is given as \(w_{uv}=\gamma A_{uv}\).
We want to define a chain here:
from epipack import MatrixEpiModel
N = 4
nodes = list(range(N))
links = [ (u,u+1,1.0) for u in range(N-1) ]
base_compartments = list("SIR")
compartments = [
(node, comp) for node in nodes for comp in base_compartments
]
model = MatrixEpiModel(compartments)
See that instead of using strings for compartments, we're using a tuple that contains the node and the compartment type. This works because tuples are hashable.
Now we want to define our reaction processes
infection_rate = 3
recovery_rate = 1
mobility_rate = 0.05
quadratic_processes = []
linear_processes = []
for node in nodes:
quadratic_processes.append(
( (node, "S"), (node, "I"), infection_rate, (node, "I"), (node, "I") ),
)
linear_processes.append(
( (node, "I"), recovery_rate, (node, "R") )
)
for u, v, w in links:
for C in base_compartments:
linear_processes.extend([
( (u, C), w*mobility_rate, (v, C) ),
( (v, C), w*mobility_rate, (u, C) ),
])
See that due to symmetry, we have to set transition reactions for \(u\rightarrow v\) as well as \(v\rightarrow u\) for the same link.
Finally, we pass the processes to the model to be converted
model.set_processes(quadratic_processes+linear_processes)
All that's left to do is to define initial conditions. Let's say we do this by assuming 20 percent of the population on the first node is infected.
initial_conditions = { ( node, "S" ): 1.0 for node in nodes }
initial_conditions[(nodes[0], "S")] = 0.8
initial_conditions[(nodes[0], "I")] = 0.2
model.set_initial_conditions(initial_conditions,allow_nonzero_column_sums=True)
Note that we pass the allow_nonzero_column_sums=True flag to suppress a warning
that the initial conditions do not sum up to unity.
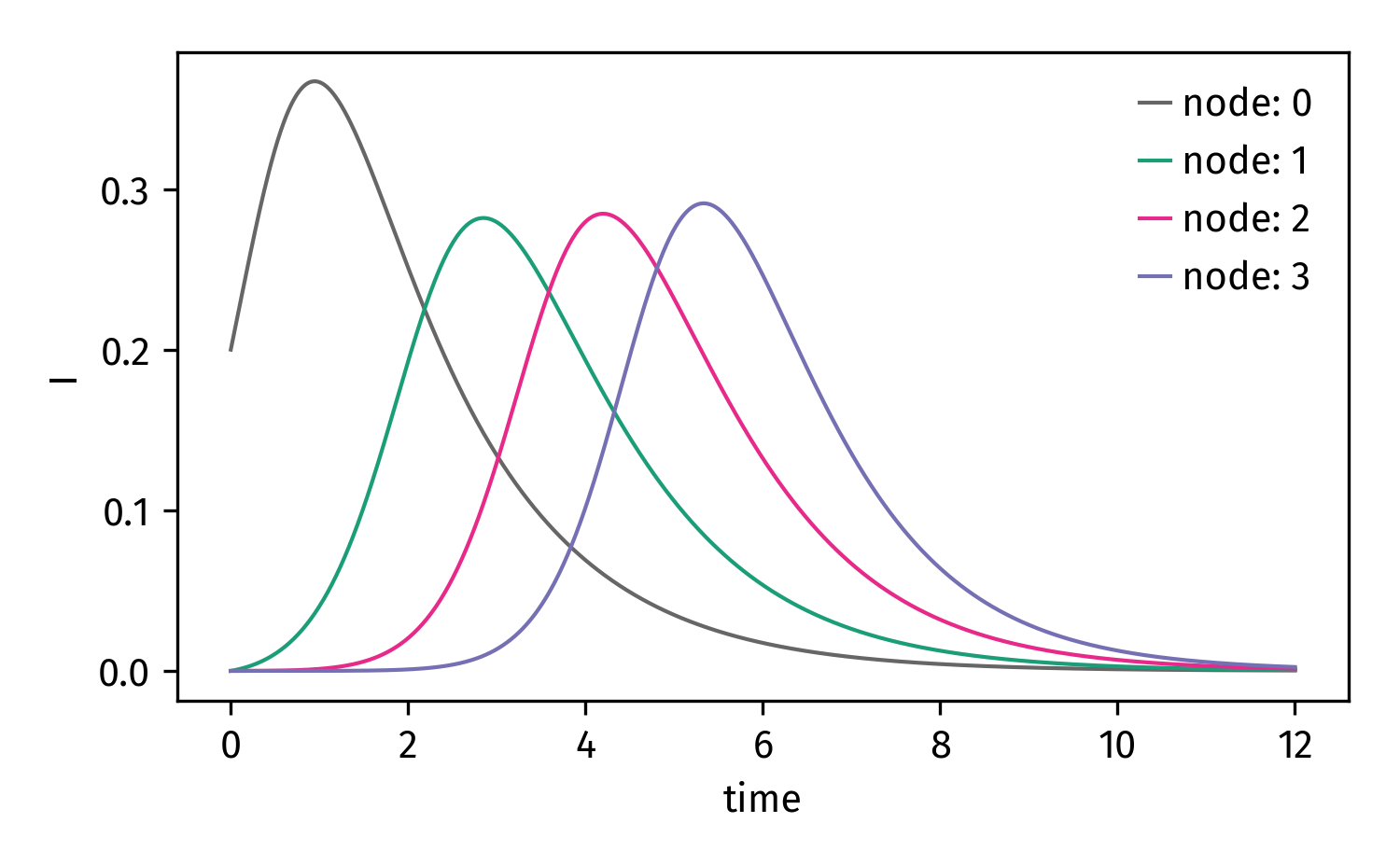
Now we can integrate and plot the I compartment for each node
# set compartments for which you want to obtain the
# result
plot_compartments = [ (node, "I") for node in nodes ]
# integrate
import numpy as np
t = np.linspace(0,12,1000)
result = model.integrate(t,return_compartments=plot_compartments)
# plot result
import matplotlib.pyplot as plt
plt.figure()
for (node, _), concentration in result.items():
plt.plot(t, concentration, label=str(node))
plt.xlabel("time")
plt.ylabel("I")
plt.legend()
plt.show()
Finally:
Note
As stated above, nodes are assumed to carry uniform equilibrium density of random walkers (as is the case in edge-centric random walks).
If, instead, you want to assume that nodes have an equilibrium density proportional to their total in-/outflux (weighted degree), you have to rescale the links (which are then equal to transition probabilities and asymmetrical).
For further info, see the supplementary material of https://science.sciencemag.org/content/342/6164/1337.figures-only
Visualization¶
Reaction-diffusion systems are interesting to watch unfold. Because we know this,
epipack provides a visualization function for reaction diffusion systems:
epipack.vis.visualize_reaction_diffusion().
Let's create a modular hierarchical network from the cMHRN package and style it
import networkx as nx
import netwulf as nw
import cMHRN
# load edges from txt file and construct Graph object
N, edges = cMHRN.fast_mhrn(8,3,7,0.18,True)
G = nx.Graph()
G.add_edges_from(edges)
# visualize and save visualization
network, config = nw.visualize(G)
nw.save("MHRN.json",network,config)
# load visualization
network, config, _ = nw.load("MHRN.json")
# get the network properties
N = len(network['nodes'])
nodes = list(range(N))
links = [ ( link['source'], link['target'], 1.0 ) for link in network['links'] ]
Subsequently, we set up the model exactly as above.
One thing that you should know is that internally, an instance of
MatrixEpiModel creates a one-dimensional \(N\times C\)-long
vector that contains the state of each compartment. In order
for the visualization function to know which entry it should plot for each node,
we have to provide it with the appropriate compartments. To this end, we construct
a list of N entries, each entry i maps node i to the compartment whose
concentration is supposed to be shown for this node.
Here, we want the node to be scaled according to its "I" compartment,
which we expect to take values between 0 and 0.3:
node_compartments = [ (node, "I") for node in nodes ]
value_extent = [0,0.3]
And finally, we can start the visualization
from epipack.vis import visualize_reaction_diffusion
dt = 0.04
visualize_reaction_diffusion(model,
network,
dt,
node_compartments,
value_extent=value_extent,
)
Lattices¶
Sometimes, a network is not the structure you want to be looking at. That's fine, you do you, go for the lattice. However, what is a lattice but a network? If you want so simulate/visualize on a lattice, simply do everything as described above, but use a lattice network
from epipack.vis import visualize_reaction_diffusion, get_grid_layout
from epipack import get_2D_lattice_network
N_side = 30
N = N_side**2
nodes = range(N)
links = get_2D_lattice_network(N)
network = get_grid_layout(N)
Now, set up everything as above and simulate on a lattice:
visualize_reaction_diffusion(model,
network,
dt,
node_compartments,
value_extent=value_extent,
config = {
'draw_nodes_as_rectangles': True,
'draw_links': False,
}
)